Autores:
Dra. Leticia Verónica Gadea Rodríguez
Hospital Pereira Rossell
Montevideo, Uruguay
Dra. Anabel Lizardi
Hospital Pereira Rossell
Montevideo, Uruguay
Dr. Pedro Andrés García Bayce
Hospital Pereira Rossell
Montevideo, Uruguay
Los autores autorizan la publicación de este caso clínico en Sociedad Latinoamericana de Radiología Pediátrica.
1. BREVE HISTORIA CLÍNICA
6 años, sexo masculino, dolor abdominal recurrente, sin otros antecedentes personales
a señalar. Consulta en la emergencia por dolor abdominal y vómitos. Del examen físico se
destacan manchas melánicas en los labios y dolor a la palpación difusa abdominal. Expulsa materias y gases.
Las imágenes 1, 2 y 3 se realizaron en el flanco izquierdo.
La imagen 4 en fosa ilíaca izquierda.
2. Hallazgos radiológicos
¿Cómo describiría los hallazgos?
3. DESCRIPCIÓN DE LOS HALLAZGOS
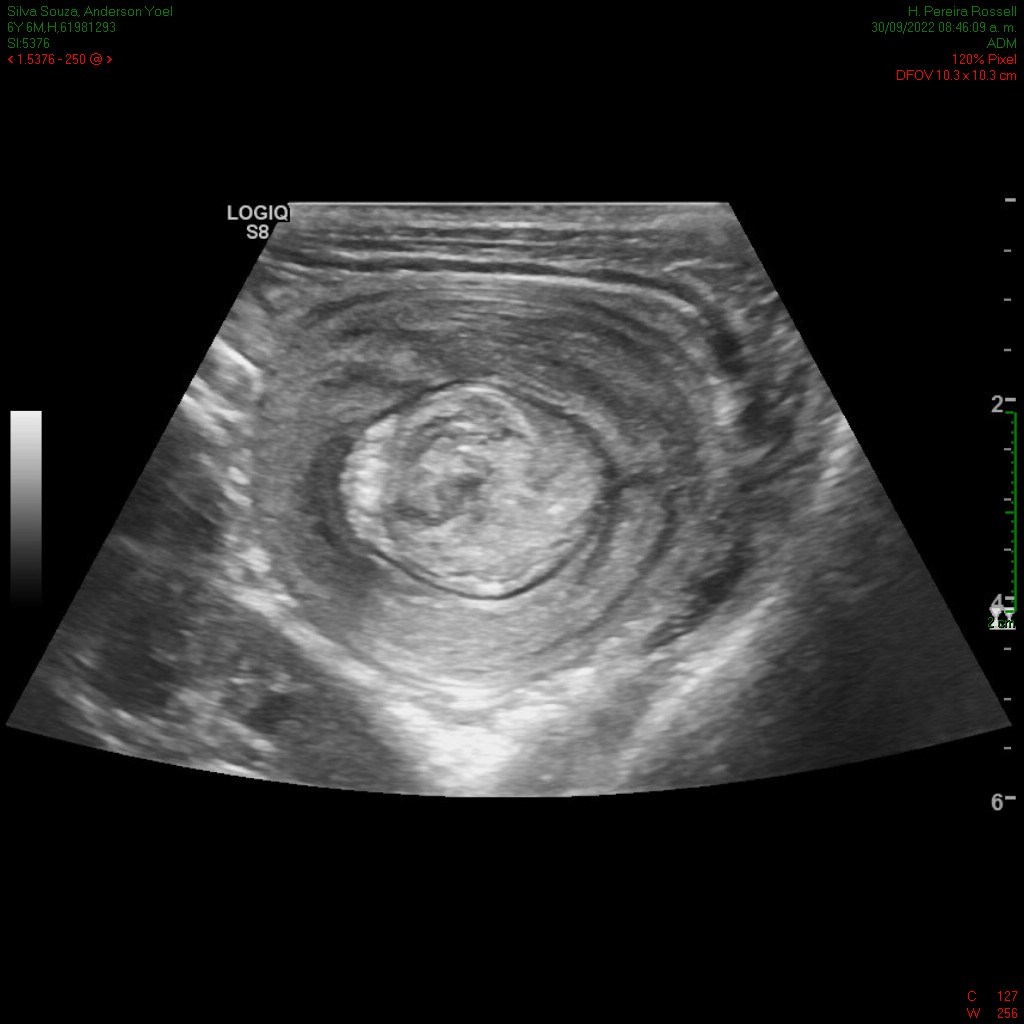
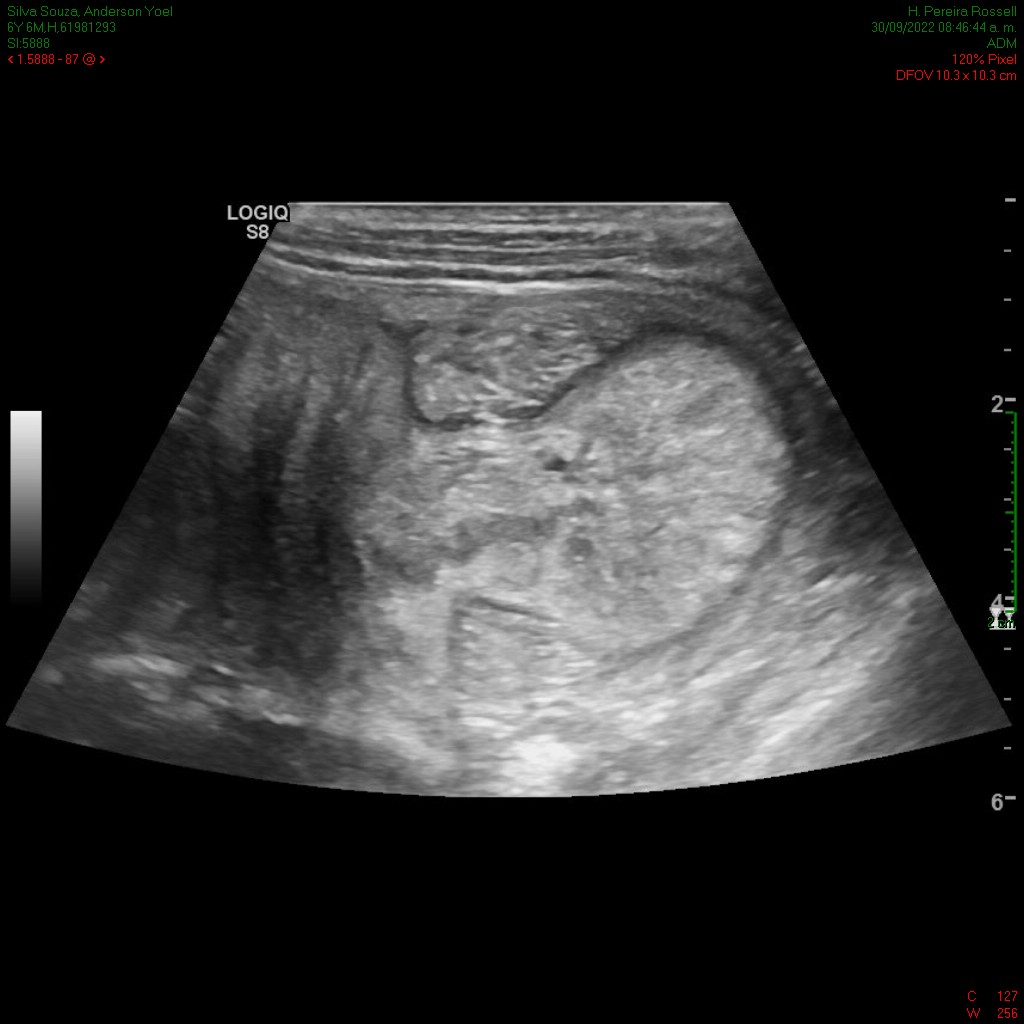
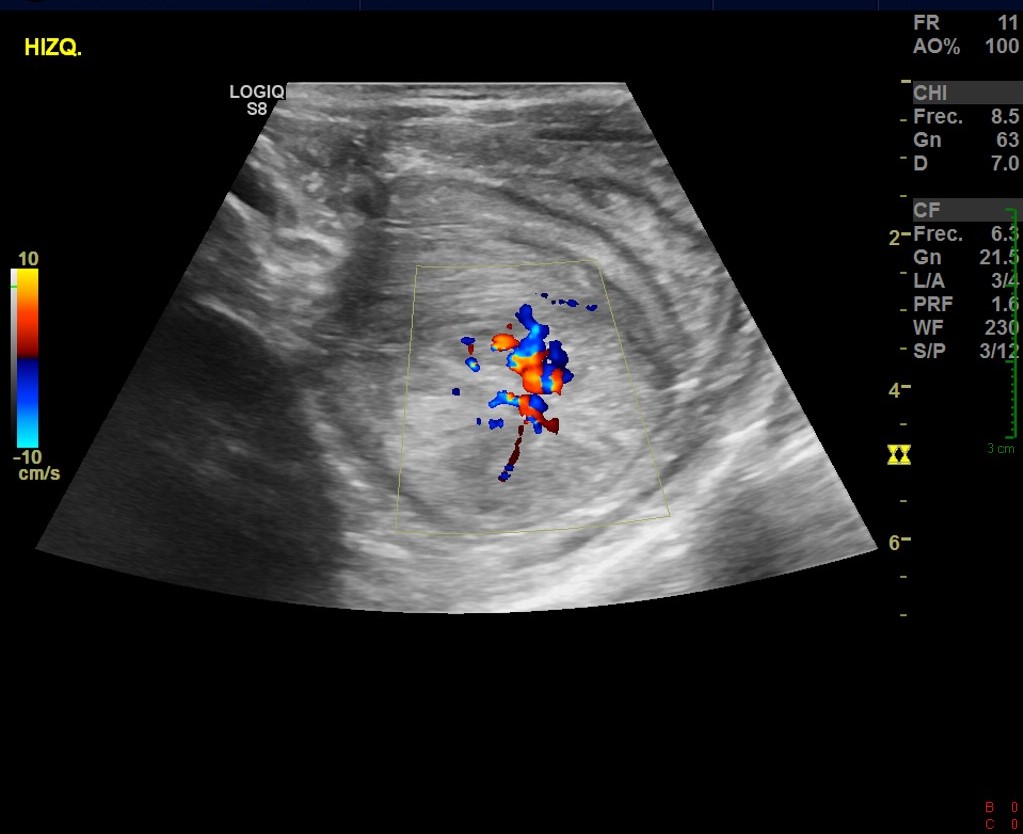
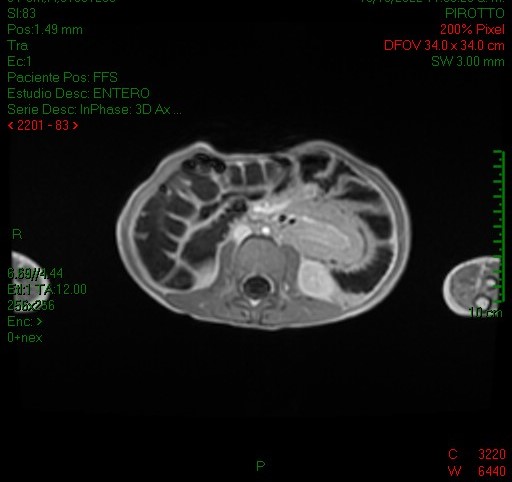
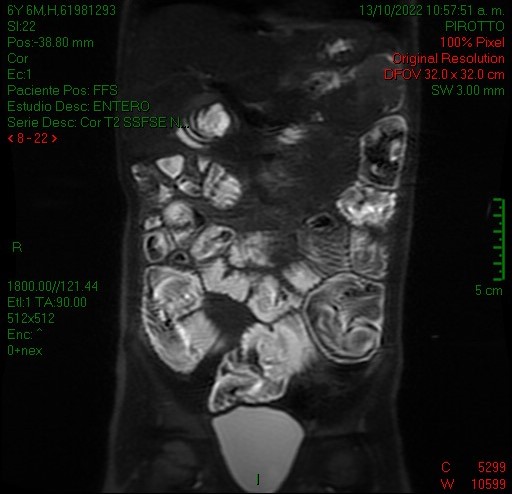
En el ultrasonido se identifica imagen con el aspecto en diana, a expensas de varias capas concéntricas de diferente ecogenicidad (imagen 1) que en el corte longitudinal (imagen 2) presenta el signo del seudorriñón compatible con invaginación intestinal.
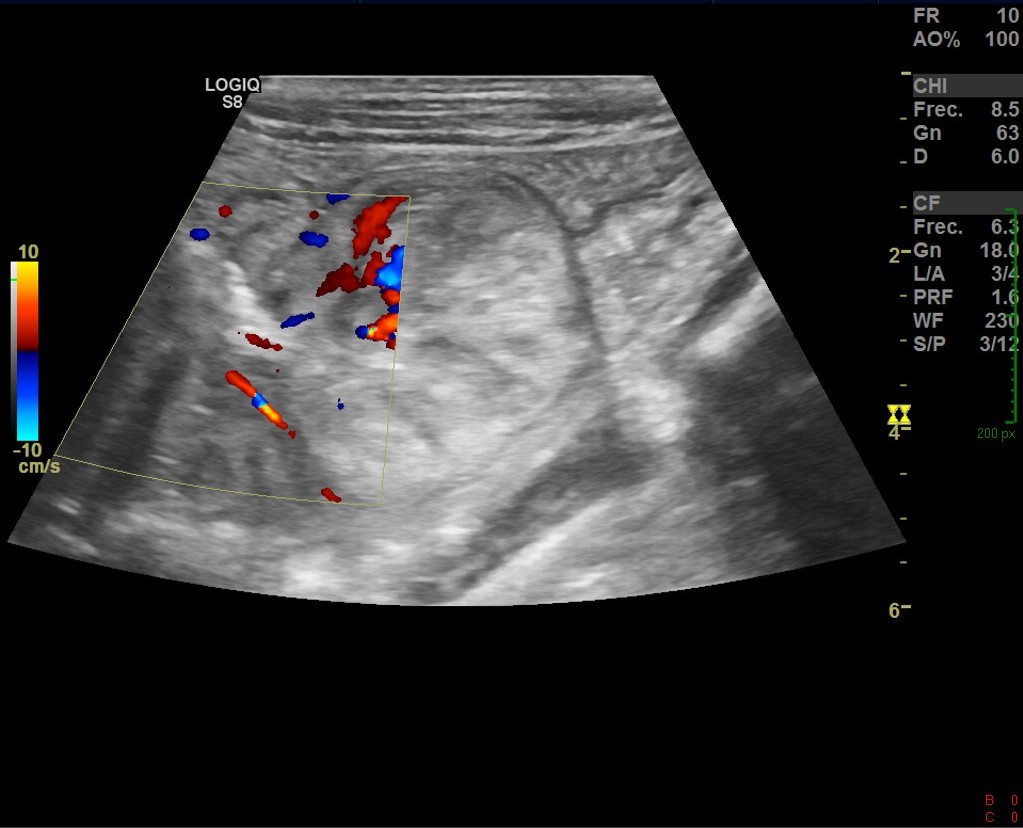
Se reconoce en el contenido de la invaginación intestinal una imagen polipoidea pediculada (imagen 3), así como otra ubicada en el lúmen de asas delgadas, localizada distalmente en la fosa ilíaca izquierda (figura 4).
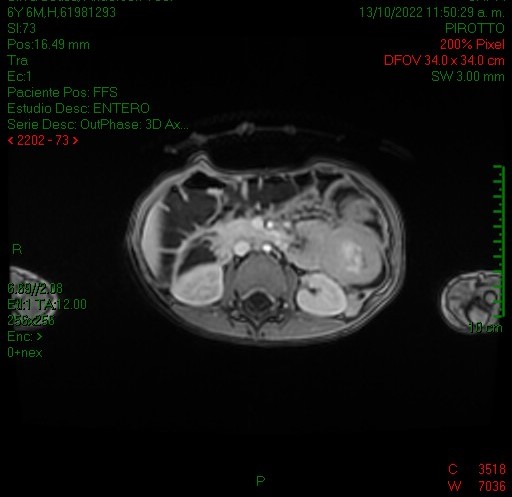
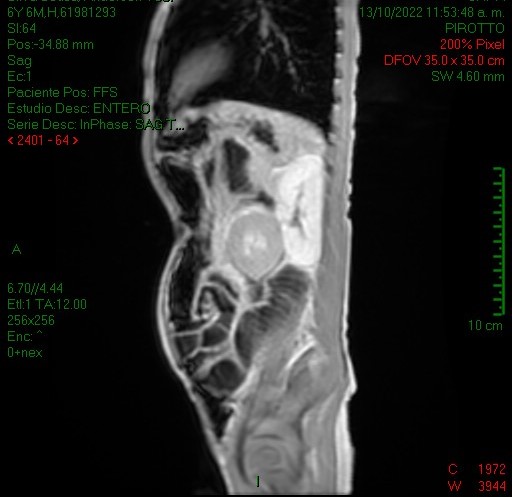
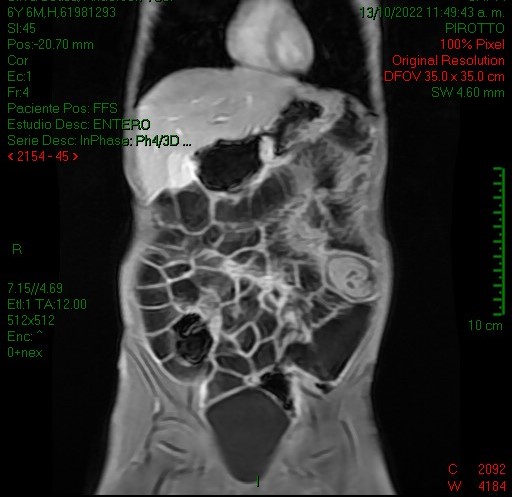
La entero resonancia realizada a este paciente demostró similares hallazgos en cuanto a la invaginación intestinal: imágenes 5, 6 y 7 axiales y sagital T1 con contraste, comprobándose la localización en asas yeyunales y caracterizándose otros pólipos, los de mayor tamaño en asas yeyunales distales a la invaginación (imagen 7 y 8, secuencias coronales T2 y T1).
4. Diagnóstico diferencial
Seleccione la opción correcta
5. Descripción del diagnóstico diferencial y de todo el caso de forma más detallada
El hallazgo de múltiples pólipos en el tubo digestivo es infrecuente en la edad pediátrica, nos debe poner en sospecha de un síndrome de poliposis intestinal, la mayoría de carácter hereditario, destacándose la importancia de que asocian un riesgo mayor del desarrollo de neoplasias malignas.
Además, estas lesiones pueden ser causa de varias complicaciones, como la invaginación intestinal vista en este paciente.
Desde el punto de vista de la imagen, es importante definir la caracterización y la localización de los pólipos en vistas al tratamiento y porque además puede ayudar a orientar al clínico en la búsqueda del diagnóstico definitivo.
El síndrome de poliposis juvenil, de herencia autosómico dominante, tiene una incidencia muy baja, para su diagnóstico debe evidenciarse 5 pólipos juveniles colorrectales, pólipos juveniles en el tracto gastrointestinal e historia familiar de poliposis juvenil.
El síndrome Bannayan-Riley-Ruvalcaba (B-R-R) causado por la mutación del gen PTEN, localizado en el cromosoma 10q23.2, se caracteriza por la asociación de múltiples lipomas, macrocefalia, hemangiomas, retraso del crecimiento y máculas hiperpigmentadas en los genitales masculinos. Entre el 35-45% de los casos, presentan pólipos intestinales, sin degeneración maligna, que pueden localizar en cualquier tramo del tracto gastrointestinal, aunque son más frecuentes en el colon y el recto.
El síndrome de Cowden, también conocido como síndrome de hamartomas múltiples, es una enfermedad de origen genético de herencia autosómico dominante que se caracteriza por triquelomas faciales, queratosis acra, pápulas o papilomas, hamartomas benignos y macrocefalia. Se presenta de forma más frecuente en mujeres de raza caucásica y se asocia a cáncer de mama, tiroides, endometrio, adenocarcinoma colorrectal, linfoma no Hodking y melanoma, entre otros.
El síndrome de Peutz-Jeghers es una enfermedad hereditaria autosómica dominante poco frecuente, caracterizada por el desarrollo de pólipos en el tubo digestivo, pigmentación mucocutánea y riesgo de desarrollar neoplasias malignas. Los pólipos son múltiples y pueden afectar a todo el tracto intestinal, desde el esófago hasta e recto. El cuadro clínico se corresponde con un paciente anémico con cuadros de invaginación recidivante.
Poliposis adenomatosa familiar (PAF) es un trastorno hereditario autosómico dominante en el que se desarrollan múltiples pólipos adenomatosos premalignos en el colon.
Las manifestaciones clínicas se suelen presentar alrededor de la segunda década de la vida y la malignización suele ocurrir en la tercera década.
6. Diagnóstico final
Haz click aquí para ver el diagnóstico final
Síndrome de Peutz-Jeghers
7. Bibliografía
1) C. Wei, W. Dayong, J. Liqun, W. Xiaoman, W. Yu, Q. Xiaohong. Colorectal polyps in children: a retrospective study of clinical features the value of ultrasonography in their diagnosis. J Pediatr Surg, 47 (2012), pp. 1853-1858.
2)M. Palacios, A. Bautista. Síndromes de poliposis intestinales. Anales de Pediatría continuada (2014).183-190.